
Properties¶
Static properties describe measurements of thermodynamic properties, spatial structure and organization
Potential/kinetic energy, heat capacity, pressure, temperature
Radial distribution functions
Number of neighbors
Order
Dynamic properties describe the movements of atoms
Diffusion coefficients
Conductivity
Time-dependent spectroscopy
Viscosity
Let us make another trajectory of our soft-sphere system:
import numpy as np
import matplotlib.pyplot as plt
from scipy.spatial import Voronoi, voronoi_plot_2d
from copy import deepcopydef periodic(coor, ndim, l):
for i in range(ndim):
if coor[i] >= 0.5*l[i]:
coor[i] -= l[i]
elif coor[i] < -0.5*l[i]:
coor[i] += l[i]
return coor
def periodic_all(coors, n, ndim, l):
for i in range(n):
coors[i]=periodic(coors[i], ndim, l)
return coors
def soft_sphere(rij_squared):
rij_squared_inv = 1/rij_squared
rij_six_inv = rij_squared_inv**3
f_val = 48 * rij_six_inv * rij_squared_inv * (rij_six_inv - 0.5)
e_val = 4 * rij_six_inv * (rij_six_inv - 1) + 1
return f_val, e_val
def compute_forces(coors, n, ndim, rcut_squared, l):
accels = np.zeros((n, ndim))
sum_pot = 0
sum_vir = 0
n_pairs = 0
for i in range(n-1):
for j in range(i+1, n):
n_pairs += 1
vect_rij = periodic(coors[i]-coors[j], ndim, l)
rij_squared = np.sum(vect_rij**2)
if rij_squared < rcut_squared:
f_val, e_val = soft_sphere(rij_squared)
accels[i] += f_val * vect_rij
accels[j] -= f_val * vect_rij
sum_pot += e_val
sum_vir += f_val * rij_squared
return accels, sum_pot, sum_vir, n_pairs
def compute_forces_cell_subdivision(coors, n, ndim, rcut_squared, l, lcell, ncell):
accels = np.zeros((n, ndim))
sum_pot = 0
sum_vir = 0
cells = np.array((coors+l/2)//lcell,'int')
cells = [(c[0],c[1]) for c in cells]
n_pairs = 0
for mx in range(ncell[0]):
for my in range(ncell[1]):
mxp1=int((mx+1)%ncell[0])
myp1=int((my+1)%ncell[1])
mym1=int((my-1)%ncell[1])
cond = [(mxp1, mym1), (mxp1,my), (mxp1,myp1), (mx, myp1)]
idxs_center = [idx for idx, c in enumerate(cells) if c==(mx,my)]
idxs_surr = [idx for idx, c in enumerate(cells) if c in cond]
for ii,i in enumerate(idxs_center):
for j in idxs_center[ii+1:]+idxs_surr:
n_pairs += 1
vect_rij = periodic(coors[i]-coors[j], ndim, l)
rij_squared = np.sum(vect_rij**2)
if rij_squared < rcut_squared:
f_val, e_val = soft_sphere(rij_squared)
accels[i] += f_val * vect_rij
accels[j] -= f_val * vect_rij
sum_pot += e_val
sum_vir += f_val * rij_squared
return accels, sum_pot, sum_vir, n_pairs
def compute_forces_neighbor_list(coors, n, ndim, rcut_squared, l, lcell, ncell, refresh, rcut_dr_squared, neighbors):
accels = np.zeros((n, ndim))
sum_pot = 0
sum_vir = 0
n_pairs = 0
if refresh:
cells = np.array((coors+l/2)//lcell,'int')
cells = [(c[0],c[1]) for c in cells]
for mx in range(ncell[0]):
for my in range(ncell[1]):
mxp1=int((mx+1)%ncell[0])
myp1=int((my+1)%ncell[1])
mym1=int((my-1)%ncell[1])
cond = [(mxp1, mym1), (mxp1,my), (mxp1,myp1), (mx, myp1)]
idxs_i = [idx for idx, c in enumerate(cells) if c==(mx,my)]
idxs_j = [idx for idx, c in enumerate(cells) if c in cond]
for ii,i in enumerate(idxs_i):
for j in idxs_i[ii+1:]+idxs_j:
n_pairs += 1
vect_rij = periodic(coors[i]-coors[j], ndim, l)
rij_squared = np.sum(vect_rij**2)
if rij_squared < rcut_dr_squared:
if i<j:
neighbors[i, j] = True
else:
neighbors[j, i] = True
else:
if i<j:
neighbors[i, j] = False
else:
neighbors[j, i] = False
if rij_squared < rcut_squared:
f_val, e_val = soft_sphere(rij_squared)
accels[i] += f_val * vect_rij
accels[j] -= f_val * vect_rij
sum_pot += e_val
sum_vir += f_val * rij_squared
refresh=False
return accels, sum_pot, sum_vir, n_pairs, neighbors, refresh
for i in range(n-1):
for j in range(i+1, n):
if not neighbors[i, j]:
continue
n_pairs += 1
vect_rij = periodic(coors[i]-coors[j], ndim, l)
rij_squared = np.sum(vect_rij**2)
if rij_squared < rcut_squared:
f_val, e_val = soft_sphere(rij_squared)
accels[i] += f_val * vect_rij
accels[j] -= f_val * vect_rij
sum_pot += e_val
sum_vir += f_val * rij_squared
return accels, sum_pot, sum_vir, n_pairs, neighbors, refresh
#Use adapted Leapfrog that gives velocities and positions at the same timestep
#The process then becomes 2 steps
def leapfrog_1(r, v, a, dt):
v_new = v + 0.5*dt*a
r_new = r + dt*v_new
return r_new, v_new
def leapfrog_2(v, a, dt):
v_new = v + 0.5*dt*a
return v_new
def initcoords(n, ndim, l, ucell):
coords = np.zeros((n, ndim))
i=0
if ndim==2:
for x in np.arange(-l[0]/2, l[0]/2, l[0]/ucell[0]):
for y in np.arange(-l[1]/2, l[1]/2, l[1]/ucell[1]):
coords[i] = np.array([x,y])
i+=1
if i == n:
break
return coords
def initvels(n, ndim, temp, seed):
np.random.seed(seed)
vels = np.random.random((n,ndim))-np.ones((n,ndim))*0.5
vels = vels/np.linalg.norm(vels, axis=1)[:,None]* np.sqrt(temp*ndim*(1-1/n))
vels -= np.sum(vels, axis=0)/n
return vels
def init(n, ndim, l, ucell, temp, seed):
step_count = 0
coors = initcoords(n, ndim, l, ucell)
vels = initvels(n, ndim, temp, seed)
accels = np.zeros((n, ndim))
return step_count, coors, vels, accels
def single_step(step_count, coors, vels, accels, density, n, ndim, rcut_squared, l, temp, nprint, dt, method, lcell, ncell, refresh, rcut_dr_squared, neighbors):
#step1 leapfrog
coors, vels = leapfrog_1(coors, vels, accels, dt)
coors = periodic_all(coors, n, ndim, l)
#compute forces
if method == 'all_pair':
accels, sum_pot, sum_vir, n_pairs = compute_forces(coors, n, ndim, rcut_squared, l)
elif method == 'cell_subdivision':
accels, sum_pot, sum_vir, n_pairs = compute_forces_cell_subdivision(coors, n, ndim, rcut_squared, l, lcell, ncell)
elif method == 'neighbor_list':
accels, sum_pot, sum_vir, n_pairs, neighbors, refresh = compute_forces_neighbor_list(coors, n, ndim, rcut_squared, l, lcell, ncell, refresh, rcut_dr_squared, neighbors)
else:
raise NotImplentedError("Unknown method")
#step2 leapfrog
vels = leapfrog_2(vels, accels, dt)
kin_energy = 0.5*np.sum(vels**2)/n #per atom
pot_energy = sum_pot/n #per atom
tot_energy = kin_energy + pot_energy #per atom
temp = 2*kin_energy/ndim
total_vel = np.linalg.norm(np.sum(vels,axis=0))
pressure = density*(total_vel**2 + sum_vir) / (n*ndim)
if step_count%nprint == 0:
print("%7i %7.3f %7.3f %7.3f %7.3f %7.3f %7i" % (step_count,
kin_energy,
pot_energy,
tot_energy,
temp,
pressure,
n_pairs))
step_count += 1
return step_count, coors, vels, accels, refresh, neighbors
def simulation(ucell, rcut, temp, density, dt, steps, nprint, method, dr=0.3, seed=0):
n = np.prod(ucell)
ndim = ucell.shape[0]
if ndim!=2:
raise NotImplementedError("Ndim other than 2 not implemented currently")
l = 1/np.sqrt(density)*ucell
rcut_squared = rcut**2
ncell = np.array(np.floor(l/rcut),'int')
lcell = l/ncell
refresh = True
cum_max_vels = 0
neighbors = np.zeros((n, n), dtype='bool')
rcut_dr_squared = (rcut+dr)**2
print("%7s %7s %7s %7s %7s %7s %7s" % ("Step", "E_k", "E_pot", "E_tot", "T", "P", "Eval r"))
traj=[]
traj_v=[]
step_count, coors, vels, accels = init(n, ndim, l, ucell, temp, seed)
for _ in range(steps):
if cum_max_vels > dr/(2*dt):
refresh=True
cum_max_vels = 0
step_count, coors, vels, accels, refresh, neighbors = single_step(step_count, coors, vels, accels, density, n, ndim, rcut_squared, l, temp, nprint, dt, method, lcell, ncell, refresh, rcut_dr_squared, neighbors)
cum_max_vels += np.max(np.linalg.norm(vels, axis=1))
traj.append(coors)
traj_v.append(vels)
return traj, traj_vucell = np.array([20, 20])
rcut = 2 ** (1 / 6)
temp = 1.0
density = 0.8
dt = 0.001
steps = 5000
ndim = len(ucell)
n = np.prod(ucell)
l = 1 / np.sqrt(density) * ucell
traj, traj_v = simulation(
ucell,
rcut,
temp,
density,
dt,
steps,
1000,
"neighbor_list",
) Step E_k E_pot E_tot T P Eval r
0 0.995 0.001 0.996 0.995 0.237 1849
1000 0.675 0.321 0.996 0.675 3.790 1047
2000 0.659 0.337 0.996 0.659 3.908 1027
3000 0.630 0.366 0.996 0.630 4.213 1036
4000 0.645 0.351 0.996 0.645 4.109 1043
Static properties¶
Static properties can be computed directly from trajectories:
Straightforward: Homogeneous, stationary (equilibrated) systems, where we can average over time and space
Inhomogeneous systems: Localized measurements instead of global averages
Nonstationary over time: No long-term time averaging

Radial distribution function (pair correlation function) g(r)¶
describes the spherically averaged local organization around any given atom
with the mean number of particles between distance and , and the number density
It can be derived from a histogram of discretized pair separations, where , which is the number of atom pairs (i,j) for which
For small :
3 dimensions: with
2 dimensions: with
Characteristics:
is proportional to the probability of finding an atom in the volume element at a distance from a given atom
is the mean number of atoms in a shell of radius and thickness surrounding the atom.
Typical behavior:
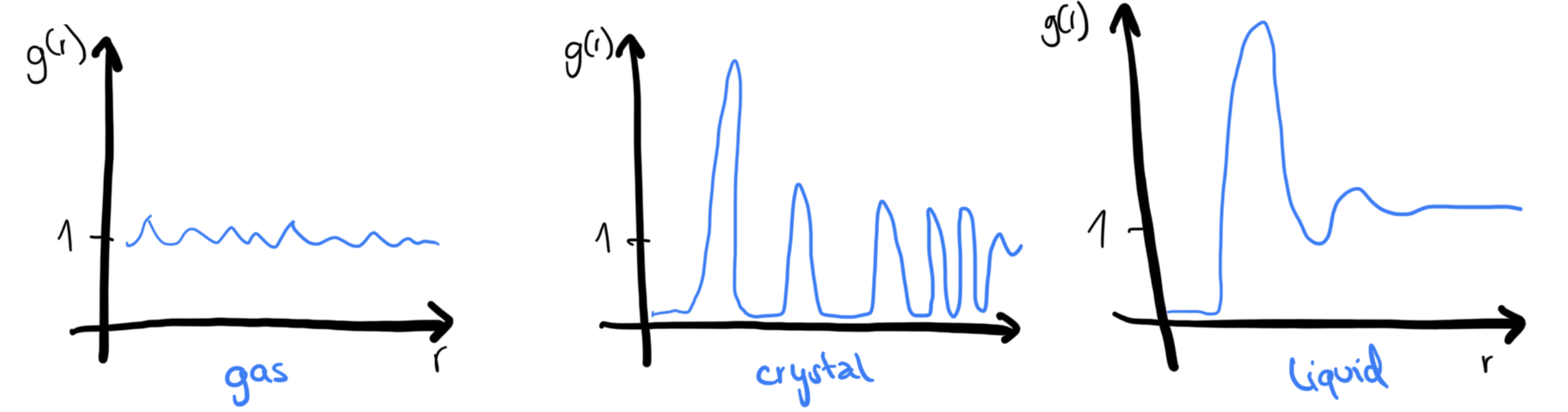
What is it used for?
central role in liquid-state physics
functions that depend on the pair separation (potential energy, pressure) can be expressed in terms of integrals involving . E.g. with the pair potential :
is related to the experimentally measurable structure factor ( is the scattering vector) by Fourier transformation It can therefore be derived experimentally from using neutron scattering or x-ray scattering data
can even be measured experimentally using microscopy for large particles
can be used to estimate errors in thermodynamic properties due to the energy cutoff. For example, for the mean potential energy: Since for large , the calculation can be simplified. For the LJ potential, it can even be evaluated analytically:
Similarly, we can compute the error in the pressure.
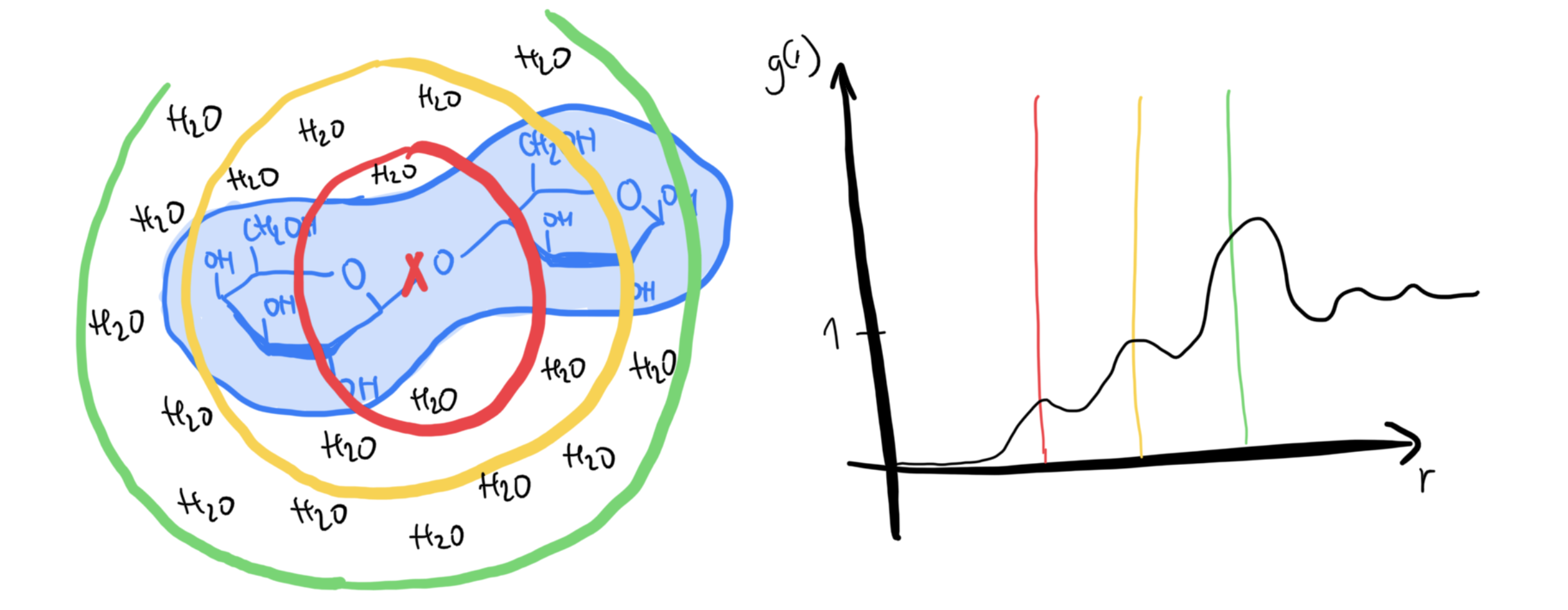
Limitations
Radial distribution functions are less meaningful for very asymmetric systems:
def compute_rdf(coords, l, dr=0.1):
n = len(coords)
density = n / np.prod(l)
ndim = len(coords[0])
rcut = min(l) / 2
n_bins = int(np.floor(rcut / dr))
print(rcut, n_bins)
h = np.zeros(n_bins)
for i in range(n - 1):
for j in range(i + 1, n):
vect_rij = periodic(coords[i] - coords[j], ndim, l)
rij = np.linalg.norm(vect_rij)
k = int(rij // dr)
if k < n_bins:
h[k] += 1
r = np.arange(0, n_bins * dr - 0.001, dr)
if ndim == 3:
g = h * 2 / n / (density * (4 / 3) * np.pi * ((r + dr) ** 3 - r**3))
else:
g = h * 2 / n / (density * np.pi * ((r + dr) ** 2 - r**2))
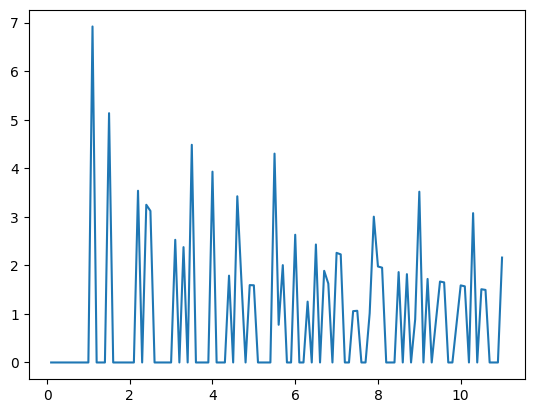
return g[1:], r[1:]i = 0
g_r, radii = compute_rdf(traj[i], [22.36067977, 22.36067977])
plt.plot(radii, g_r)11.180339885 111
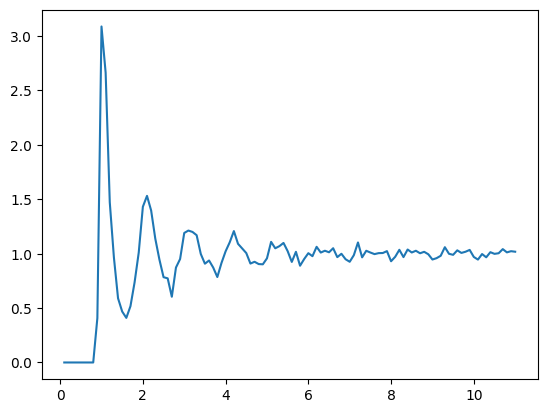
i = -1
g_r, radii = compute_rdf(traj[i], [22.36067977, 22.36067977])
plt.plot(radii, g_r)11.180339885 111
Local order¶
Local order gives information about e.g. how molecules are organized, how large the exposed surface of a large molecule is, etc.
Voronoi subdivision (= tesselation)¶
Construct polyhedra around atoms/molecules such that all points that are closer to a particular atom/molecule than any other belong to its polyhedron. Adjacent atoms/molecules then share a common face of their polyhedra.
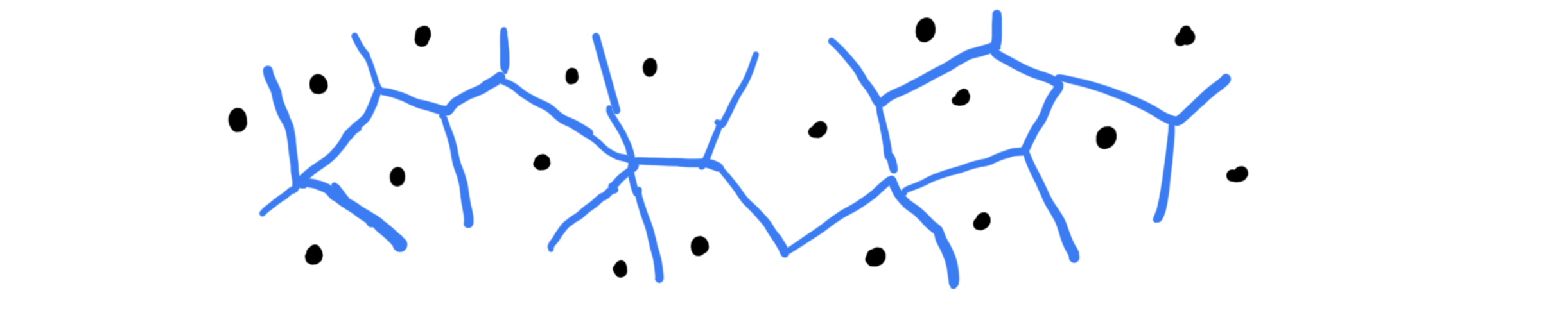
It is commonly used for:
Shape of polyhedra and their volume have a physical meaning
Accurate neighbor counts even for non-spherical molecules
Computation of solvation shells
For mixtures: Number of specific species in the x-th solvation shell
def supercell(coords, L):
new_points = []
for i in [-1, 0, 1]:
for j in [-1, 0, 1]:
if i == 0 and j == 0:
continue
for p in coords:
if p[0] > L[0] / 2 or p[0] < -L[0] / 2:
print(p)
if p[1] > L[1] / 2 or p[1] < -L[1] / 2:
print(p)
new_points.append(p + np.array([i * L[0], j * L[1]]))
return np.array(list(coords) + new_points)
l = [22.36067977, 22.36067977]
points = supercell(traj[0], l)
plt.close()
vor = Voronoi(points)
fig = voronoi_plot_2d(vor, show_vertices=False)
plt.xlim([-l[0] / 2, l[0] / 2])
plt.ylim([-l[1] / 2, l[1] / 2])
plt.show()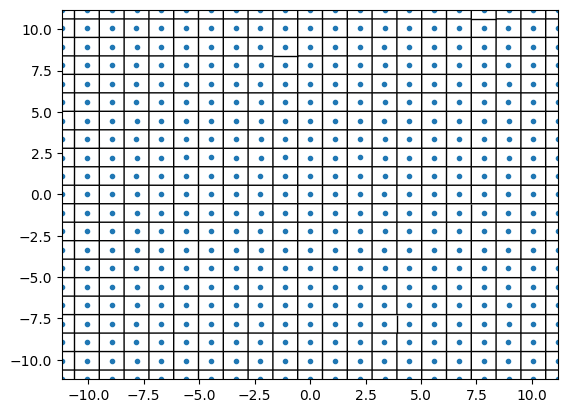
points = supercell(traj[-1], l)
plt.close()
vor = Voronoi(points)
fig = voronoi_plot_2d(vor, show_vertices=False)
plt.xlim([-l[0] / 2, l[0] / 2])
plt.ylim([-l[1] / 2, l[1] / 2])
plt.show()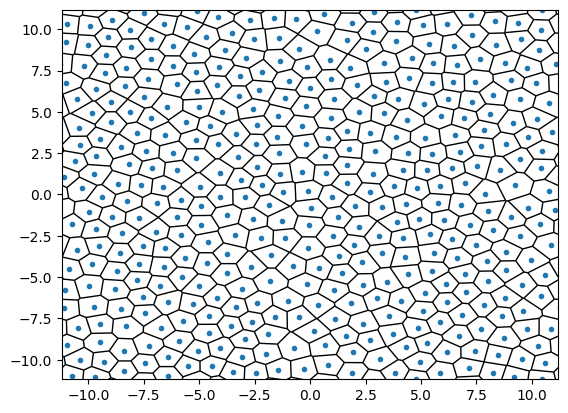
We can easily obtain neighbor counts:
_ = plt.hist([len(x) for x in vor.regions], bins=12)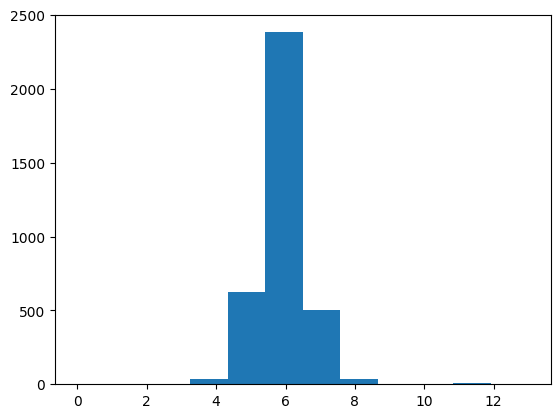
Long-range order¶
primarily addresses the local structure, and gives only little direct information on long-range (crystalline) order.
The local density at a point can be expressed as a sum over atoms:
Fourier-transforming gives
To test for long-range order, we compute , and can, for example, follow how a system solidifies.
In our simulation, the long-range order decreases over time, since we start from an ordered system that then gets more and more random:
def compute_lattice(coords, UCELL, L):
k = np.array(
[2 * np.pi * 1 * UCELL[0] / L[0], 2 * np.pi * -1 * UCELL[1] / L[1]]
)
sr = 0
si = 0
for r in coords:
t = np.dot(r, k)
sr += np.cos(t)
si += np.sin(t)
return np.sqrt(sr**2 + si**2) / len(coords)
plt.close()
x = np.arange(0, 1000, 100)
plt.plot(x, [compute_lattice(traj[i], np.array([20, 20]), l) for i in x])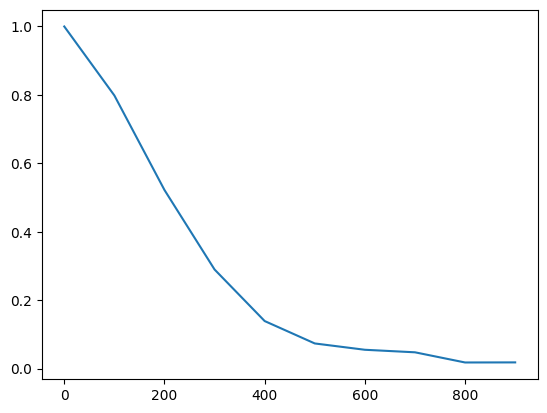
Dynamic properties¶
Dynamic properties are related to nonequilibrium properties. For example, transport coefficients like the diffusion, shear and bulk viscosity, and thermal conductivity arise from mechanical perturbations (concentration, velocity, thermal gradients).
Linear response theory relates the reaction of an equilibrium system to a small external perturbation via equilibrium correlation functions. We can thus compute dynamic properties from equilibrium trajectories.
Namely, the Green-Kubo formalism connects a macroscopic property (=response property of a system) to its equilibrium fluctuations. For example, diffusion is the response to concentration inhomogeneity.
Diffusion coefficient¶
Diffusion coefficients accessible from the mean-square displacement (\textbf{Einstein expression}):
Derivation:
Fick’s law with ... local density or concentration:
Multiply by and integrate along :
Integrate by parts and assume is normalized to 1: \hfill
Integral is an expectation value since behaves like a probability:
is linear at long time scales:
is the average over the particles:
We thus relate the miscroscopic dynamics () with a macroscopic property ()
For periodic boundary conditions, we need the true atomic displacements without periodic wraparound (we need to “unfold the trajectory”)!
Unwrapping the trajectory¶
def unwrap(traj, l, ndim):
new_traj = [traj[0]]
for i in range(1, len(traj)):
new = deepcopy(traj[i])
old = new_traj[i - 1]
change = new - old
for ii, p in enumerate(change):
for j in range(ndim):
if p[j] >= l[j] / 2:
new[ii][j] -= l[j]
elif p[j] <= -l[j] / 2:
new[ii][j] += l[j]
new_traj.append(new)
return new_traj
unfolded_traj = unwrap(traj, l, 2)Diffusion via Einstein expression¶
t0 = 1000
dts = np.arange(0, len(unfolded_traj) - t0, 10)
msd = [
np.mean(np.sum((unfolded_traj[t0 + t] - unfolded_traj[t0]) ** 2, axis=1))
for t in dts
]
plt.close()
plt.plot(dts, msd)
plt.show()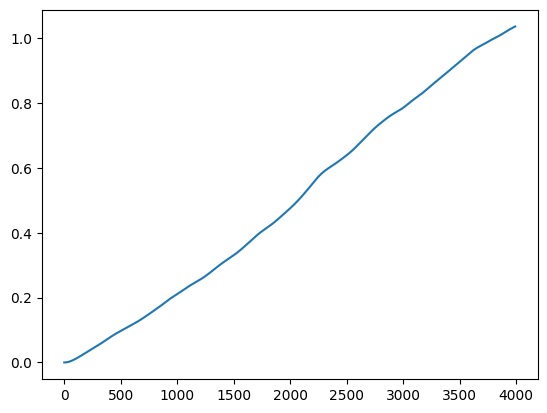
t0 = 1000
t = 3000
np.sum(np.sum((unfolded_traj[t0 + t] - unfolded_traj[t0]) ** 2, axis=1)) / (
2 * ndim * n * t * dt
)np.float64(0.06550639059451377)Diffusion via Velocity Autocorrelation Function (Green-Kubo)¶
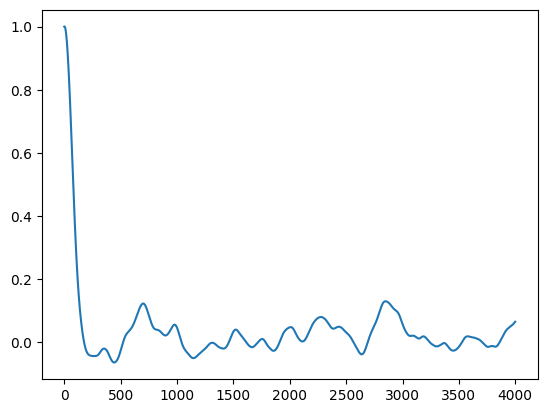
Alternative without unfolding: Green-Kubo expression based on the velocity autocorrelation function:
Derivation:
Start with: \hfill
Reformulate as \hfill
One can show that , which at turns into : \hfill
Important (for Einstein or Green-Kubo relation): For very small box-sizes, we need to include a correction factor!
t0 = 1000
t = 4000
norm = sum([np.dot(traj_v[t0][j], traj_v[t0][j]) for j in range(n)])
plt.plot(
[tt for tt in range(t)],
[
sum([np.dot(traj_v[t0 + tt][j], traj_v[t0][j]) for j in range(n)])
/ norm
for tt in range(t)
],
)
t0 = 1000
t = 3000
norm = 1
sum(
[
sum([np.dot(traj_v[t0 + tt][j], traj_v[t0][j]) for j in range(n)])
/ norm
* dt
for tt in range(t)
]
) / (ndim * n)np.float64(0.09285037676101865)Shear viscosity¶
The shear viscosity can be obtained via
where denotes a sum over the three pairs of components xy, yz and zx. It characterizes the rate at which some component (e.g. y) of momentum diffuses in a perpendicular (x) direction. This expression is unfortunately unusable with periodic boundary conditions!
Alternative: Green-Kubo expression based on the pressure tensor autocorrelation function:
with
Thermal conductivity¶
The thermal conductivity can be obtained via
where is the instantaneous excess energy of atom , is the mean energy and is a sum over vector components. This expression is unfortunately unusable with periodic boundary conditions!
Alternative: Green-Kubo expression based on the heat flux autocorrelation function:
with