-body potentials¶
Classic approach (as approximation of true, quantum-mechanical energy)}
+ ...
The best choice of potential depends on the state of the system (gas, liquid, solid), and the type of bonds (covalent bonds, non-bonded interactions, crystal/metal lattices)
For non-bonded interactions, a two-body potential is usually a good description already. For bonded interaction, many more terms are needed!
Two-body potentials ¶
Noble gases:
Lennard-Jones potential 1924: to model noble gases.
Buckingham potential 1938: allows for a more flexible form than Lennard-Jones
Covalent (only diatomic) bonds:
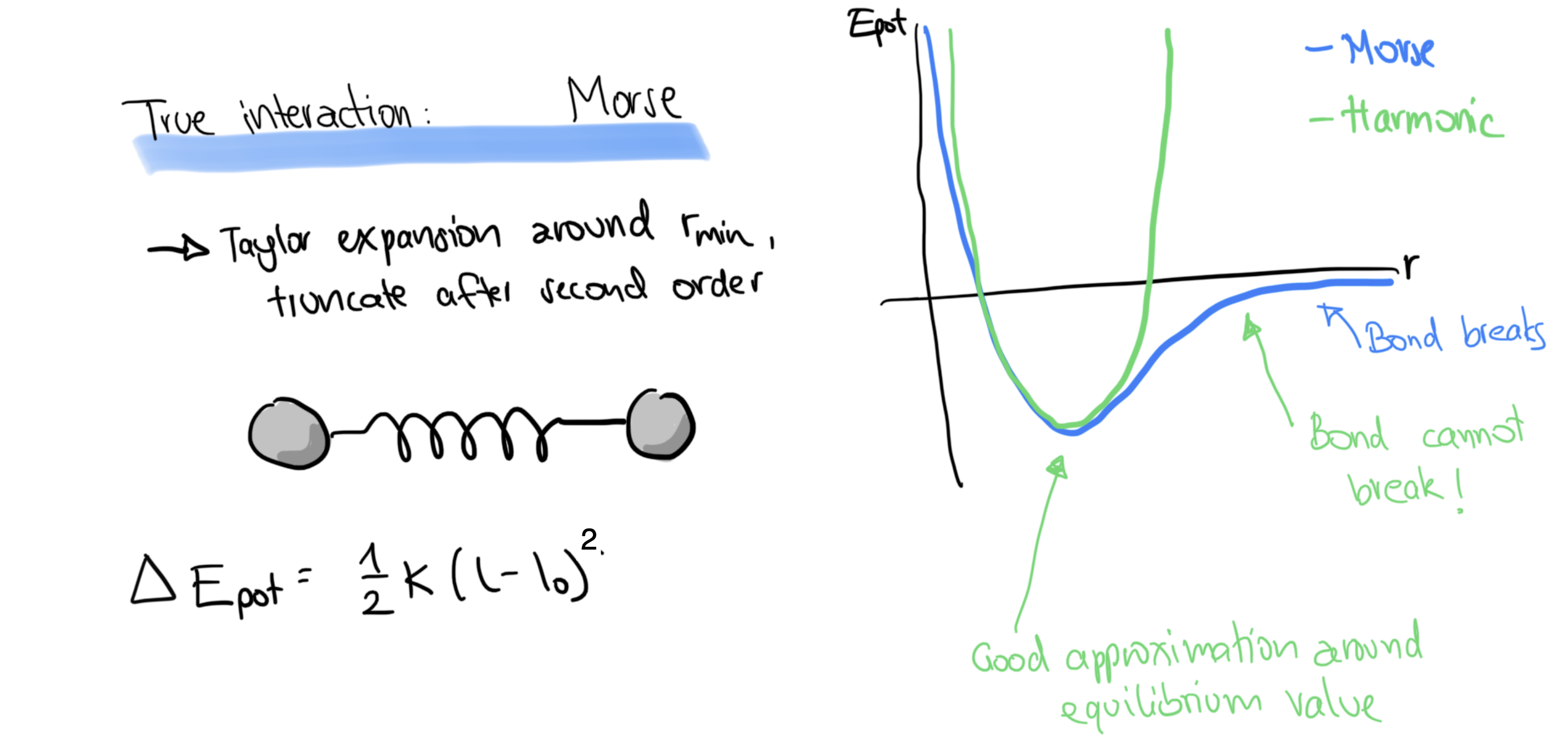
Morse potential 1929: describes many diatomic systems well. Minimum at with the potential energy
Solid state:
Stillinger-Weber potential 1985: and to model solid and liquid phases of Si, GaN, 2D materials, etc.
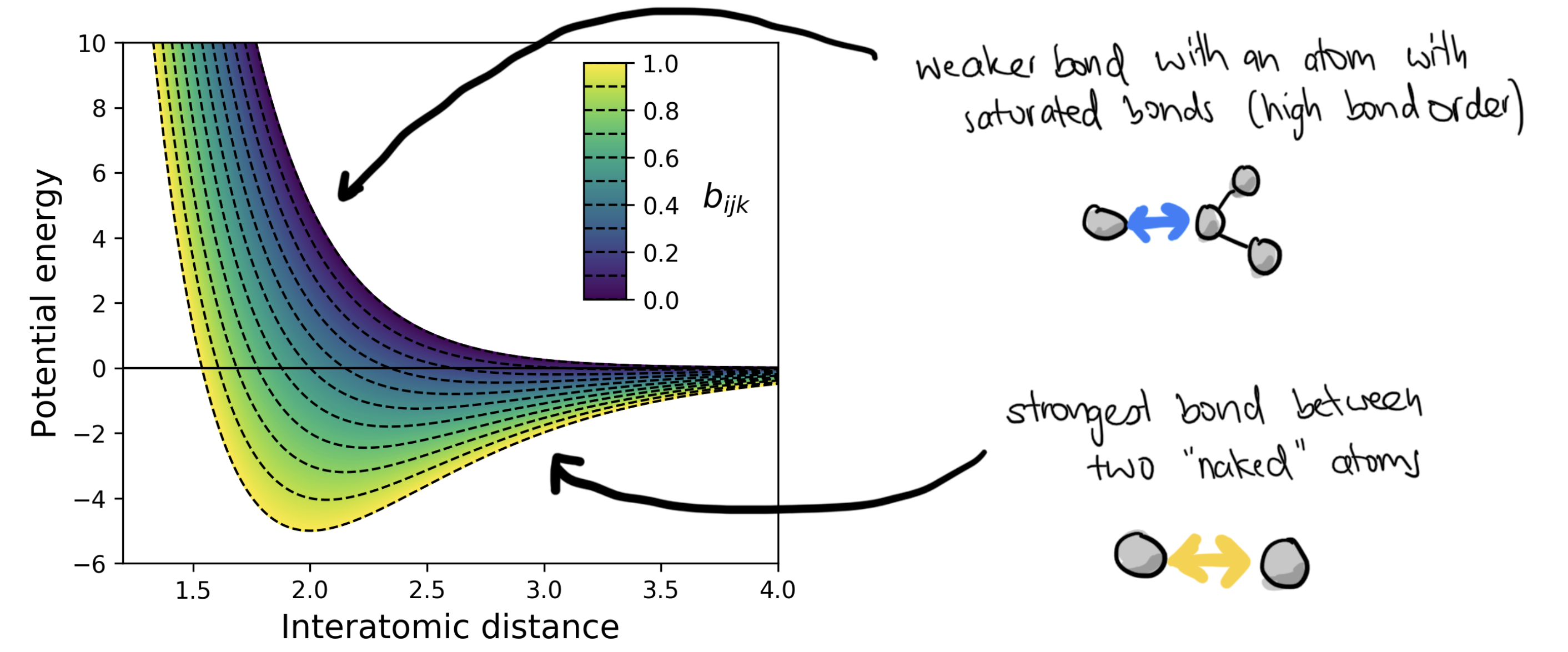
Tersoff’s bond-order potential 1988: which is a two-body potential where the attractive part is modulated by the bond order which depends on
Molecular mechanics¶
Molecular systems (with a bent toward organic molecules in fluid phases)
Potential has bonded and non-bonded interactions
Atoms have vdW radii, partial charges and possibly polarizabilities
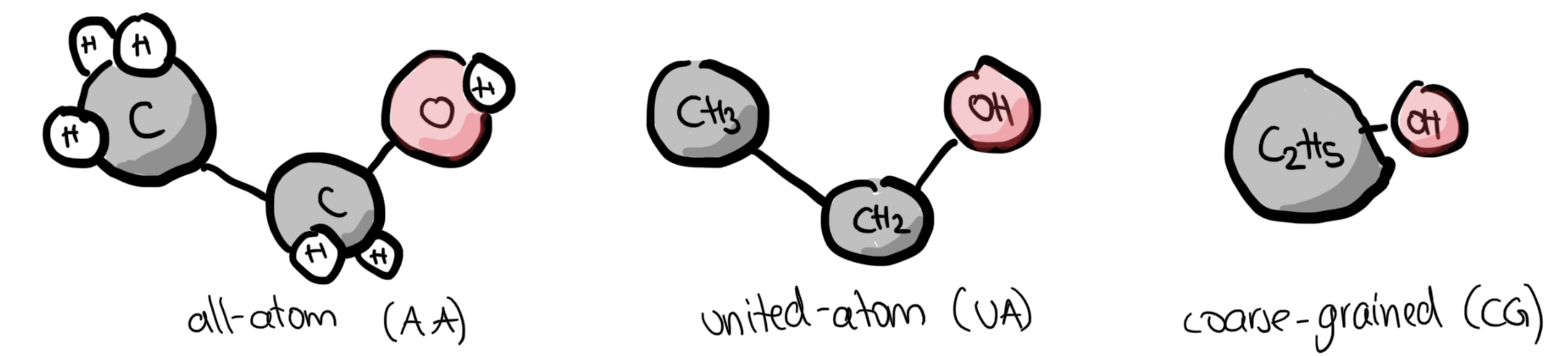
Molecules can be treated in an all-atom, united-atom (implicit hydrogen) or coarse-grained (multiple atom form a bead) fashion. Parts of the simulation can also be at the QM-level (QM-MM hybrid approach)
e.g. OPLS-AA 1996: The potential is a sum of bonded and non-bonded energy terms:
Bonded terms:
Bond-term
Angle-term
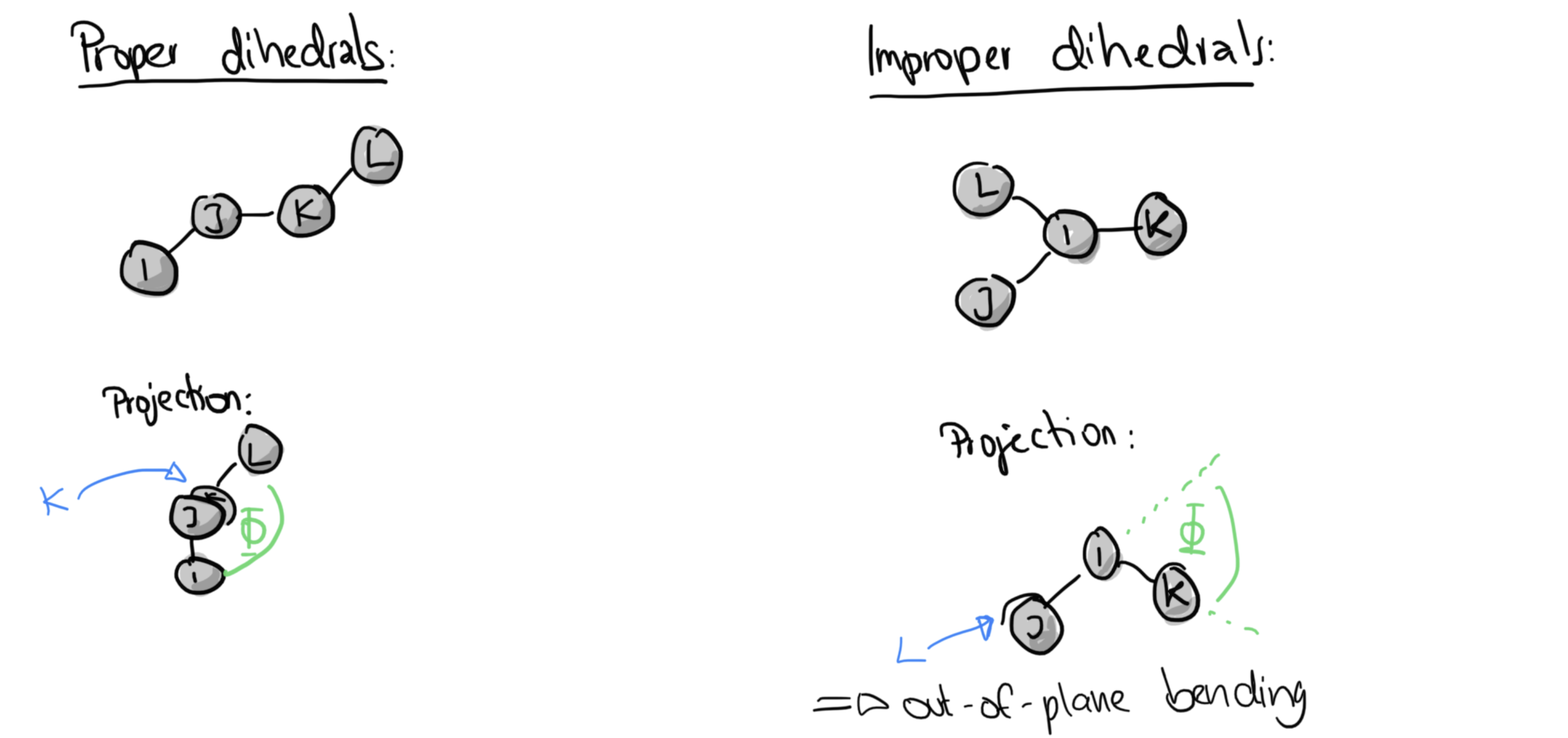
Dihedral-term
Non-Bonded terms: Combined vdW and charge interactions: with combining rules and and is 1/0.5/0 if I and J are more than/exactly/less than 3 bonds apart.
Two-body terms in molecular potentials (= bonds)¶
Three-body terms in molecular potentials (= angles)¶

Four-body terms in molecular potentials (= dihredral angles)¶
In the following, we will generate a trajectory for ethane, and try to reverse-engineer the potential:
from openmm.app import *
from openmm import *
from openmm.unit import *
import MDAnalysis as md
import MDAnalysis.transformations
import MDAnalysis.analysis
import MDAnalysis.analysis.msd as msd
import MDAnalysis.analysis.rdf as rdf
from sys import stdout
import subprocess
import numpy as np
import matplotlib.pyplot as pltsubprocess.run(["wget", "https://raw.githubusercontent.com/hesther/teaching/main/scm_exercise4/ethane.pdb"],stdout = subprocess.DEVNULL,stderr = subprocess.DEVNULL)
subprocess.run(["wget", "https://raw.githubusercontent.com/hesther/teaching/main/scm_exercise4/ethane.xml"],stdout = subprocess.DEVNULL,stderr = subprocess.DEVNULL)
CompletedProcess(args=['wget', 'https://raw.githubusercontent.com/hesther/teaching/main/scm_exercise4/ethane.xml'], returncode=0)pdb = PDBFile('ethane.pdb')
forcefield = ForceField('ethane.xml')
system = forcefield.createSystem(pdb.topology, nonbondedMethod=NoCutoff, constraints=HBonds)
integrator = LangevinIntegrator(298.15*kelvin, 5.0/picoseconds, 2.0*femtoseconds)
integrator.setConstraintTolerance(1e-5)
simulation = Simulation(pdb.topology, system, integrator)
simulation.context.setPositions(pdb.positions)
print('Minimizing...')
st = simulation.context.getState(getPositions=True,getEnergy=True)
print(F"Potential energy before minimization is {st.getPotentialEnergy()}")
simulation.minimizeEnergy(maxIterations=100)
st = simulation.context.getState(getPositions=True,getEnergy=True)
print(F"Potential energy after minimization is {st.getPotentialEnergy()}")
print('Equilibrating...')
simulation.reporters.append(app.StateDataReporter(stdout, 500, step=True,
potentialEnergy=True, temperature=True, separator=','))
simulation.context.setVelocitiesToTemperature(150.0*kelvin)
simulation.step(2500)
print('Running Production...')
# Clear simulation reporters
simulation.reporters.clear()
# Reinitialize simulation reporters
simulation.reporters.append(app.StateDataReporter(stdout, 5000,
step=True, time=True, potentialEnergy=True, temperature=True,
speed=True, separator=','))
# write out a trajectory (i.e., coordinates vs. time) to a DCD file every 100 steps - 0.2 ps
simulation.reporters.append(app.DCDReporter('ethane.dcd', 100))
# run the simulation
simulation.step(100000)
Minimizing...
Potential energy before minimization is 4.902478229380711 kJ/mol
Potential energy after minimization is 4.3898291478741935 kJ/mol
Equilibrating...
#"Step","Potential Energy (kJ/mole)","Temperature (K)"
500,9.964168291421284,296.07841633076924
1000,13.603892433724699,158.16148129574543
1500,33.835767276800944,256.35367325204845
2000,26.857078433685444,191.26451993072845
2500,15.609034600938289,243.75801823514556
Running Production...
#"Step","Time (ps)","Potential Energy (kJ/mole)","Temperature (K)","Speed (ns/day)"
5000,10.000000000000009,23.50267163081441,540.2708428997199,0
10000,19.999999999999794,15.817215479691324,698.6049451979376,29.4
15000,29.99999999999425,11.578670331720101,325.8712992297204,30.2
20000,40.00000000000292,18.676473083050375,225.9719807135084,33.4
25000,50.00000000001514,35.2721186020697,277.3981626933412,36.6
30000,60.00000000002736,12.549529628162745,192.70881506693976,37.9
35000,70.00000000001826,19.499511015385792,314.6460745007441,38.8
40000,79.99999999999496,21.994178776356062,326.5993656762624,40
45000,89.99999999997165,6.9073579017079645,182.56445556391068,40.5
50000,99.99999999994834,11.62858921868106,411.78016244798755,40.8
55000,109.99999999992504,12.012523965528688,264.05024608877466,41.4
60000,119.99999999990173,8.314999146615943,307.42645264457894,41.1
65000,129.99999999989262,11.90288621320278,304.13619365651823,40.6
70000,139.99999999994037,10.081479275725624,329.8475817679048,41.3
75000,149.99999999998812,23.938675960620706,357.1241830960322,41.4
80000,160.00000000003587,20.04996457946585,300.2466752898448,40.8
85000,170.00000000008362,10.900216664059133,293.4716351587902,41.2
90000,180.00000000013137,21.11175804641629,446.9669142260622,41.5
95000,190.0000000001791,15.939904502922495,543.4425677781309,41.8
100000,200.00000000022686,15.659245875611425,573.075286421655,42
u = md.Universe("ethane.pdb", "ethane.dcd")
u.atoms.names/opt/conda/lib/python3.12/site-packages/MDAnalysis/coordinates/DCD.py:171: DeprecationWarning: DCDReader currently makes independent timesteps by copying self.ts while other readers update self.ts inplace. This behavior will be changed in 3.0 to be the same as other readers. Read more at https://github.com/MDAnalysis/mdanalysis/issues/3889 to learn if this change in behavior might affect you.
warnings.warn("DCDReader currently makes independent timesteps"
array(['C1', 'C2', 'H11', 'H12', 'H13', 'H21', 'H22', 'H23'], dtype=object)bond_length = [md.core.topologyobjects.Bond([0,1],u).value() for ts in u.trajectory]
bondcounts, binedges, otherstuff = plt.hist(bond_length, bins=120)
plt.title('C-C bond length histogram')
plt.xlabel('Bond length (Angstrom)')
plt.ylabel('Counts')
plt.show()
From the distribution of bond lengths, we can compute the potential of mean force, which should approximately recreate the potential we used to make the trajectory (namely the bonded term with nm and kJ/mol nm, values taken from the XML file above).
The potential of mean force is defined as
where if the probability (the histogram) we calculated above.
def bond(r, K, r0):
return K/2*(r-r0)**2
kB = 8.31446/1000 # Boltzmann constant in kJ/mol K
Temp = 298.15 # simulation temperature
bondcounts[bondcounts==0] = 0.1
pmf = -kB*Temp*np.log(bondcounts)
pmf = pmf - np.min(pmf)
bincenters = (binedges[1:] + binedges[:-1])/2
plt.plot(bincenters, pmf, label='potential of mean force')
plt.xlabel('Bond length (Angstrom)')
plt.ylabel('Relative free energy (kJ/mol)')
plt.title('C-C bond length pmf')
r_nm=np.arange(0.1498,0.158,0.001)
r_a=r_nm*10
plt.plot(r_a, bond(r_nm, 1945727.36, 0.15380), label='harmonic potential')
plt.legend()
plt.show()
Let us also analyze the H-C-C-H torsion. To do so, we only need to change md.core.topologyobjects.Bond to md.core.topologyobjects.Dihedral and use the correct indices.
phi = [md.core.topologyobjects.Dihedral([2,0,1,5],u).value() for ts in u.trajectory]
phicounts, phi_binedges, otherstuff = plt.hist(phi, bins=120)
plt.title('H-C-C-H dihedral angle histogram')
plt.xlabel('Phi (degree)')
plt.ylabel('Counts')
plt.show()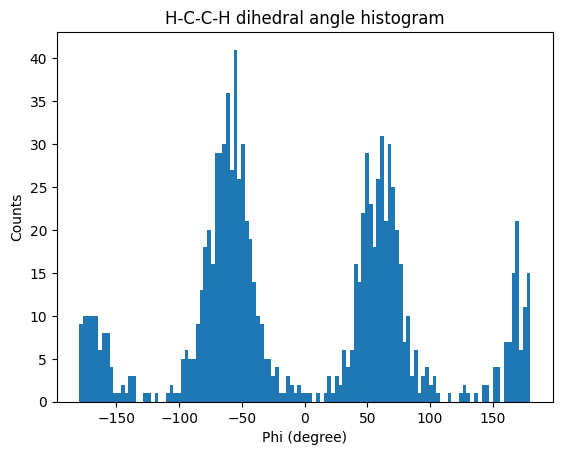
To compare with the term, we have to remember that there are nine terms that change when we turn along the C-C bond (below, we only followed how a single bond changed, and thus only one term in the energy was affected).
H11-C1-C2-H21
H11-C1-C2-H22
H11-C1-C2-H23
H12-C1-C2-H21
H12-C1-C2-H22
H12-C1-C2-H23
H13-C1-C2-H21
H13-C1-C2-H22
H13-C1-C2-H23
We therefore multiply a single term by 9 to get a good comparison:
def dihedral(phi, K, phi0, n):
return K*(1+np.cos(n*phi-phi0))
kB = 8.31446/1000 # Boltzmann constant in kJ/mol K
Temp = 298.15 # simulation temperature
phicounts[phicounts==0] = 0.1
pmf = -kB*Temp*np.log(phicounts)
pmf = pmf - np.min(pmf)
phi_bincenters = (phi_binedges[1:] + phi_binedges[:-1])/2
plt.plot(phi_bincenters, pmf, label='potential of mean force')
plt.title('H-C-C-H torsion pmf')
plt.xlabel(r'$\phi$ (rad)')
plt.ylabel('Relative free energy (kJ/mol)')
phi_rad=np.arange(-np.pi, np.pi, 0.1)
phi_deg = phi_rad*180/np.pi
plt.plot(phi_deg, dihedral(phi_rad, 0.5021, 0, 3)*9, label='harmonic potential')
plt.legend()
plt.show()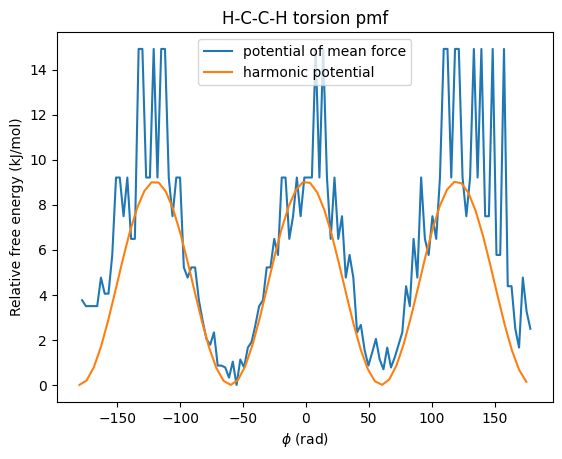
Partial charges¶
There is no physical quantity such as an “atomic charge”, there are therefore many valid schemes to assign partial charges, e.g. (among many others)
Formal charges
Restrained fit to the electrostatic potential (RESP)
Mulliken charges
Bader charges
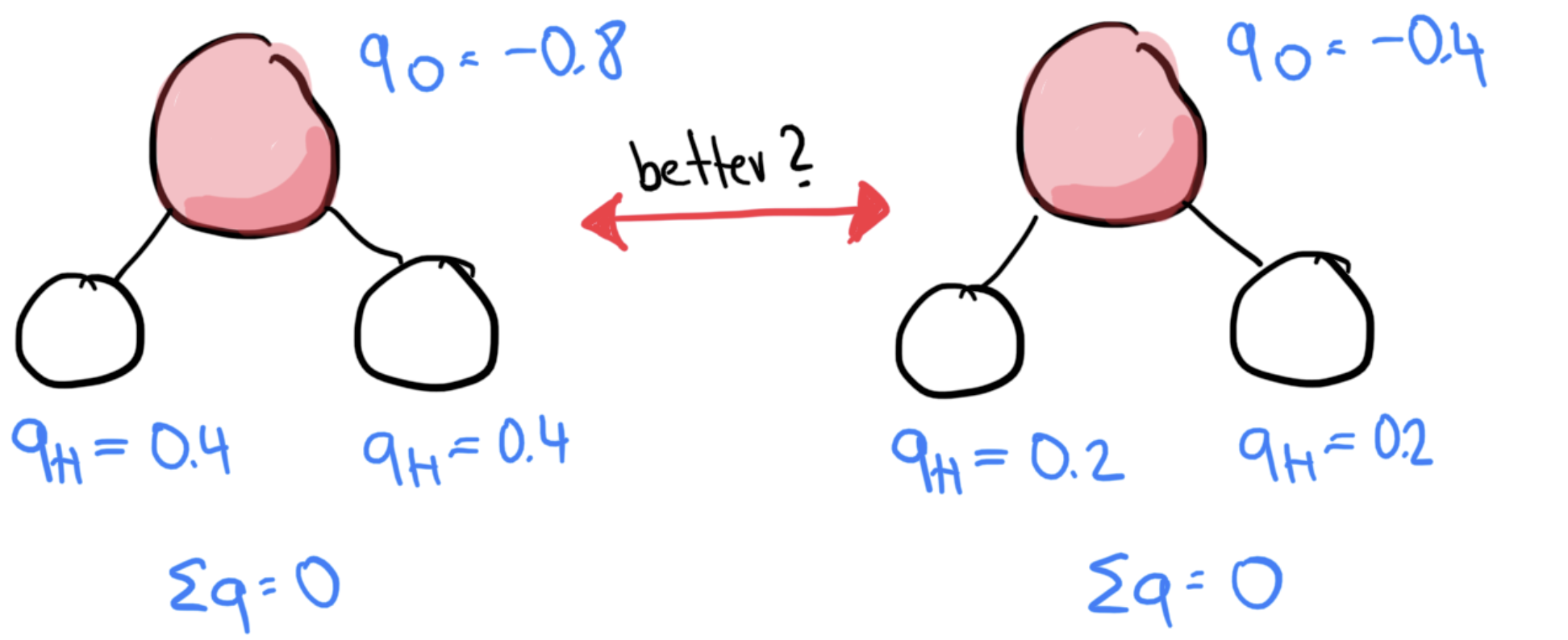
Polarizability¶
Reactive (molecular) force fields¶
For LJ, Buckingham, Morse-type FFs, reactivity is not a problem:
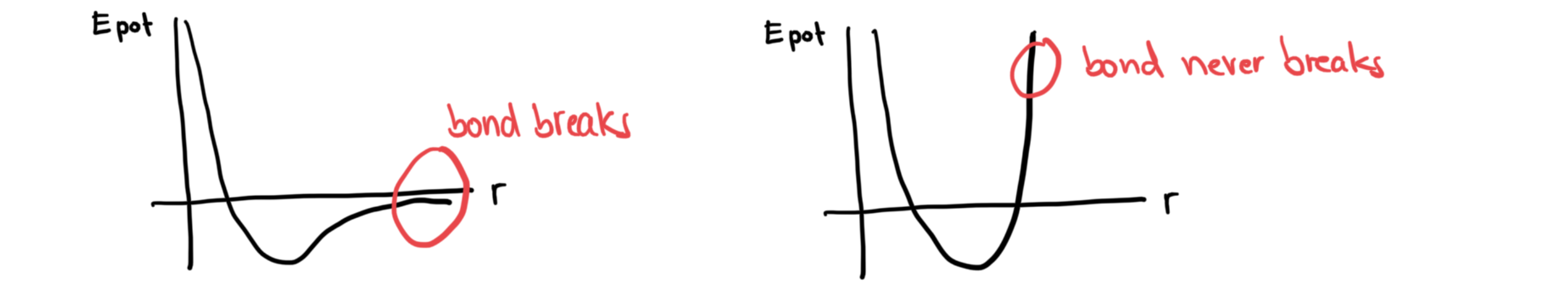
But for molecular force fields, because we change the Morse term to an harmonic term, all bonds become unbreakable
Possible solutions:
General reactive force fields based on bond-order potentials, for example ReaxFF
Force fields without enforced functional form, e.g. from machine learning
For simple cases, e.g. protonation reactions, one can also work with ghost atoms
A potential for water¶
There are nearly 50 water potentials reported in literature, which follow the general forms
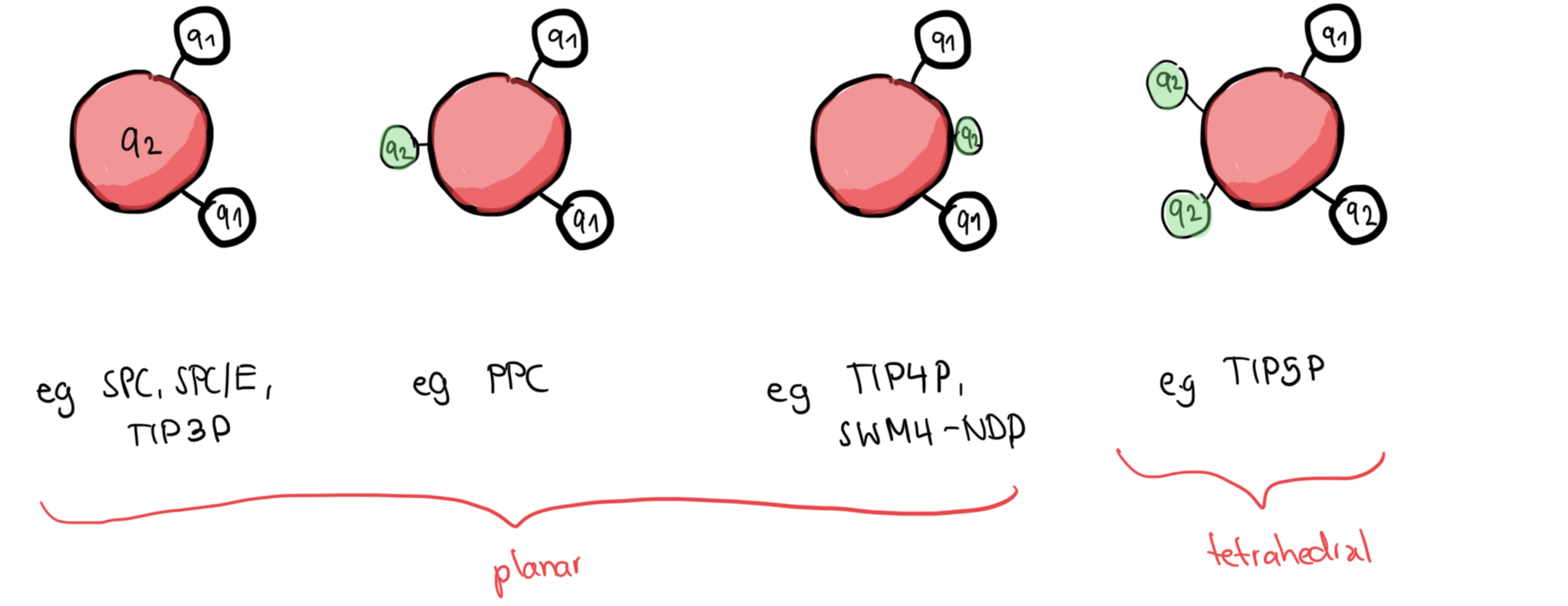
Yet, none of them capture all the properties of water well:
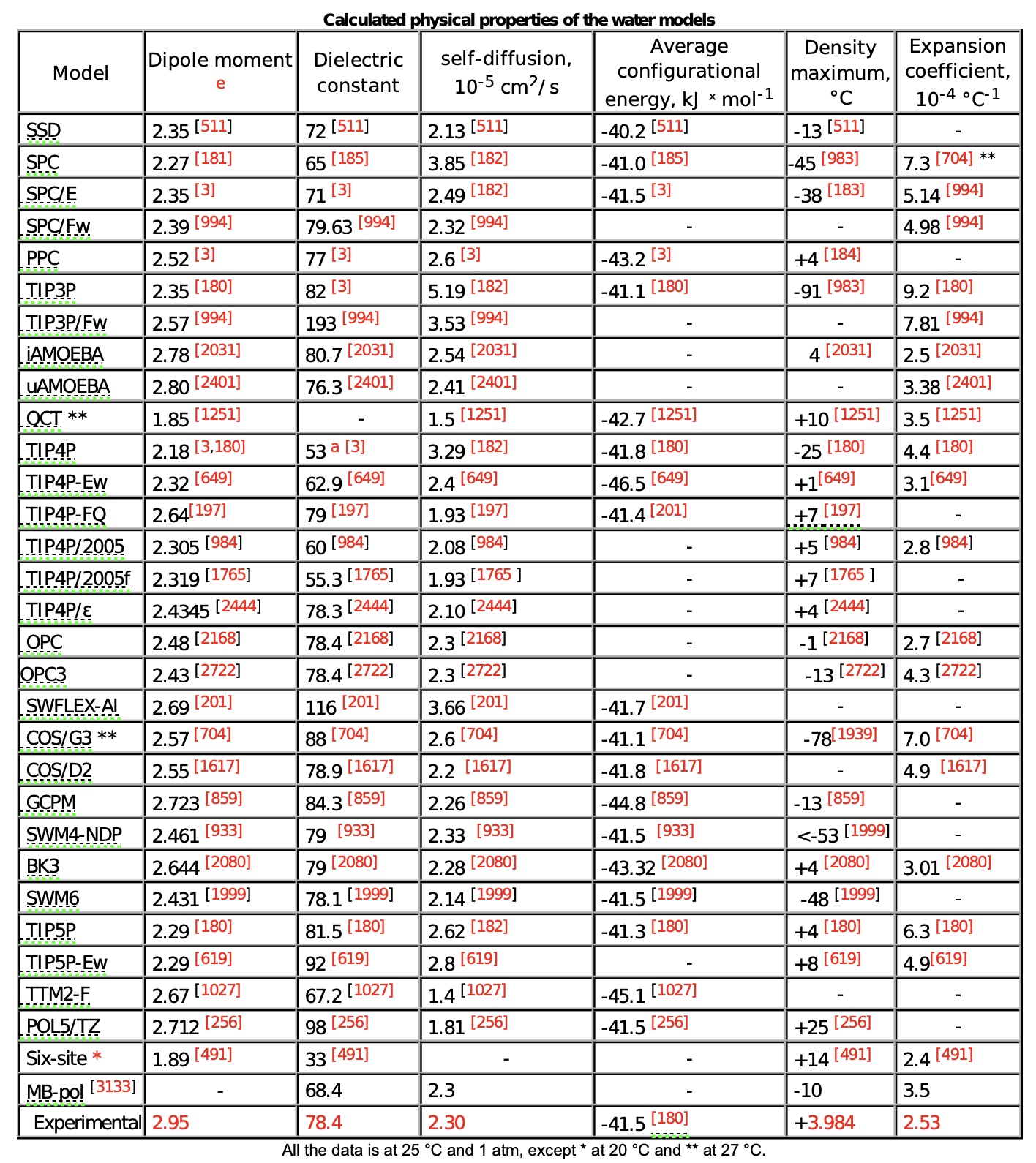 (from https://
(from https://